Das chemfig Paket kann dazu genutzt werden um einfache zweidimensionale chemische Strukturen und Schemata in LaTeX zu setzen. [1] Zum zeichnen der Moleküle und der Strukturen wird TikZ verwendet. Das Paket selbst wird in einigen anderen Chemiepaketen wie carbohydrates, mychemistry und substances benötigt beziehungsweise von diesen genutzt.
Das Paket wird mit \usepackage{chemfig} eingebunden. Falls es nach dem Einbinden des Paketes zu Fehlermeldungen kommt, ist eine häufige Ursache dafür, dass die TikZ Variante auf dem entsprechenden Rechner zu alt ist. Wobei dieser Fehler nur noch selten auftritt.
Der Basisbefehl heißt wie das Paket selbst \chemfig den folgenden Aufbau:
\chemfig[Option]{Code} beziehungsweise etwas ausführlicher \chemfig[Liste mit Komma getrennten Key = Value Paaren]{Code des Moleküls}
Über die Optionen des Befehls können unter anderem die Verbindungen der nachfolgenden Objekte festgelegt beziehungsweise verändert werden.
Im Fall von zwei Elementen (Atomen, Gruppen, Knoten etc.) lässt sich die Syntax des chemfig Befehls vereinfacht in dieser Form darstellen.
\chemfig{<Element 1><Art der Bindung>[<Winkel>,<Koeffizenten>,<ggf. zusätzlicher TikZ Code>]<Element 2>}
| Eingabe | Ausgabe |
| \chemfig{A-B} |  |
| \chemfig{A=B} | 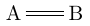 |
| \chemfig{A~B} | 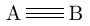 |
| \chemfig{A>B} |  |
| \chemfig{A<B} |  |
| \chemfig{A>:B} | 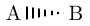 |
| \chemfig{A<:B} | 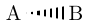 |
| \chemfig{A>|B} | 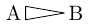 |
| \chemfig{A<|B} | 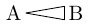 |
Die Gestaltung der Bindungen können verändert werden.
Wenn der vertikale Abstand innerhalb einer doppelt beziehungsweise einer dreifach Bindung verändert werden soll erfolgt dies über den Befehl \setdoublesep{Hoehe}, der Standardwert beträgt 2pt.
Der horizontale Abstand zwischen zwei Elementen beträgt standardmäßig 3em und lässt sich mit \setatomsep{Laenge} anpassen.
Der horizontale Abstand zwischen Bindung und Element, Standardwert 2pt, kann mit dem Befehl \setbondoffset{Laenge} verändert werden.
Mit \setbondstyle{TikZ Code} lassen sich Stiländerungen wie im folgenden Beispiel \setbondstyle{line width = 1pt, red} umsetzen. Soll wieder auf die ursprünglichen Einstellungen zurück gewechselt werden, reicht es aus den setbond Befehl mit einem leeren Argument beziehungsweise ohne Parameter \setbondstyle{} zu setzen.
Es existieren drei Arten von Winkeln: gegebene, absolute und relative Winkel.
Bei den vorgegeben Winkeln ist eine Schrittweite von +45 Grad pro Schritt als Standard vorgegeben. Die Schritte gehen von der waagerechten als Basis aus. \chemfig{A-[Zahl von 0 bis n]B}
Die mögliche Anzahl der verschiedenen Winkel ist von der Schrittweite abhängig, da sich die Winkel ab einer gewissen Zahl wiederholen.
| Zahl | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ... |
| Winkel | 0° | 45° | 90° | 135° | 180° | 225° | 270° | 315° | 360° | ... |
Da 360°/45° = 8 ist, gibt es in der Standardeinstellung acht verschiedene unterscheidbare Winkeleinstellungen.
Mit dem Befehl \setangleincrement{Grad-Zahl} kann die Schrittweite verändert werden. Womit sich auch die Anzahl der unterscheidbaren Winkelstellungen verändert.
Die absoluten und relativen Winkel werden mit einem vorangestellten Doppelpunkt (:) beziehungsweise zwei Doppelpunkten (::) genutzt.
\chemfig{A-[:Winkel] B} Absolut
\chemfing{A-[::Winkel] B} Relativ
Der Unterschied zwischen den beiden Varianten zeigt sich bei der Drehung.| \chemfig{[:60]H-[:30]O-[:-30]H} | vs. | \chemfig{[:60]H-[::30]O-[::-60]H} |
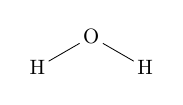 | vs. | 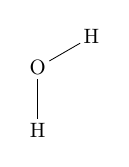 |
\chemfig{A-[Zahl Winkel, Koeffizient]B}
Eingabe:
\chemfig{H-[:30,2]O-[:-30]H}
Ausgabe: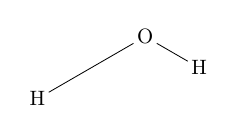
\chemfig{H-C(-[2]H)(-[6]H)-C(-[2]H)(-[6]H)-O-[1]H}
Ausgabe: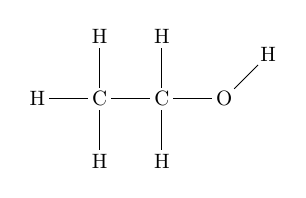
Es lassen sich sowohl vollständige wie auch unvollständige Ringe mit und ohne Atome oder Molekülen darstellen. Der Aufbau ist dabei wie folgt:
<Atom>*<Anzahl>(<Code>)| Eingabe | Ausgabe |
| \chemfig{C*6(-C=C-C=C-C=)} | 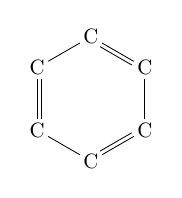 |
| \chemfig{*6(-=-=-=-)} | 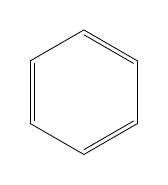 |
| \chemfig{C*6((-O-N=H_2)=-=-=-)} | 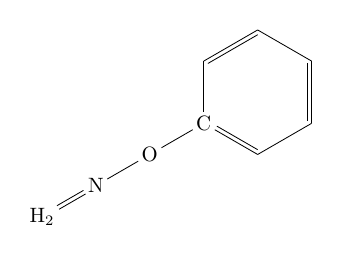 |
| \chemfig{C*6(-C=C-C=C-C=)} | 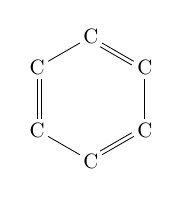 |
| \chemfig{C*6(-C=C-C=C-)} | 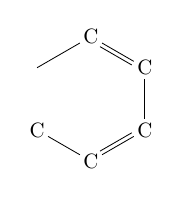 |
| \chemfig{*6(-=-=-)} | 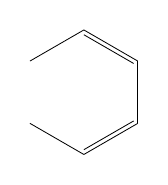 |
Innerhalb der zwei Befehle \schemestart und \schemestop können Schemata erstellt werden. Mit dem Befehl \chemname kann dies beschriftet werden.
\schemestart
\chemname[Dimension]{...}{...}
\schemestop
Die Option Dimension legt den Abstand zwischen der Zeichnung der Beschriftung fest.
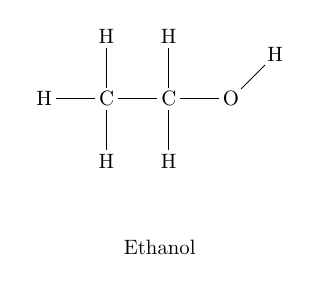
\schemestart
\chemname[8ex]{\chemfig{H-C(-[2]H)(-[6]H)-C
(-[2]H)(-[6]H)-O-[1]H}}{Ethanol}
\schemestop
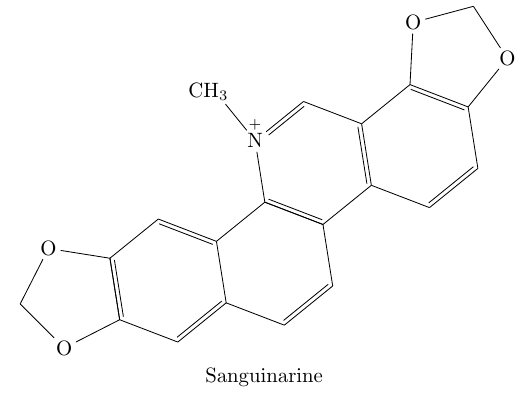
\schemestart
\chemname{
\chemfig{[:45]O*5(-*6(-=*6(-=-*6(-*6(-=-*5(-O--O-)
=-)=-=\chemabove{N}{\scriptstyle+}(-[::270]CH_3)-=)
--)-=-=)--O--)}}{Sanguinarine}
\schemestop
Die mit chemfig gesetzten Strukturen können auch in einer figure Umgebung gesetzt werden. Wenn sie innerhalb der figure Umgebung gesetzt können sie auf diese Art auch in das Abbildungsverzeichnis kommen.
\begin{figure}[!htpb]
\chemfig{[::60]N*6(=-(<:(<[::115]H)
*5(-N(-CH_3)----))=-=-)}
\caption{Nikotin}
\end{figure}
Sinnvoller ist jedoch, wenn das chemstyle Paket verwendet wird, es in ein Schemata Verzeichnis, welches von chemstyle bereit gestellt wird, einzufügen.
Mit dem Befehl lewis beziehungsweise Lewis lassen sich Valenzstriche und Elektronenformeln darstellen. Der Unterschied zwischen beiden Befehlen besteht lediglich darin, dass bei Lewis die eingefügten Symbole (Striche oder Punkte) zusammen mit dem Element in einer bounding box sind, während sie bei lewis nicht zusammen sind. Das kann insbesondere bei der Elektronenformeldarstellung zu einem Verschwimmen beziehungsweise Überlagern von Elektronenpaaren, die verschiedenen Atomen zugeordnet sind, führen. Sodass in einem solchen Fall eher Lewis anstelle von lewis verwendet werden sollte.
In den neusten Versionen des Paketes (Version 1.5, 05.03.2020) werden die Befehle Lewis und lewis durch einen neuen Befehl charge ersetzt. In zukünftigen Versionen sollen diese Befehle entfernt werden.
Der Autor des Pakets hat am Ende der offiziellen Paketbeschreibung zahlreiche Beispiele, welche mit chemfig erstellt wurden, eingefügt.